Methods
The basic concept:
This module shows the relationship between allele, haplotypes and genotypes.
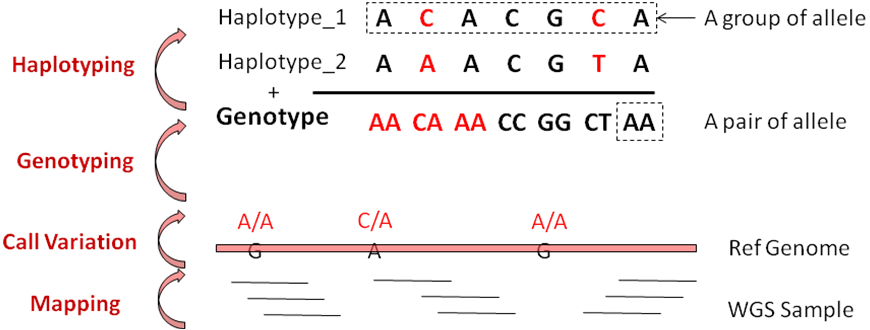
The term variant is used to refer to a specific region of the genome which differs between two genomes.
Different versions of the same variant are called alleles. When working with VCF files the term alleles = REF + ALT.
REF, reference allele that the base is found in the reference genome. ALT, alternative allele that the base is found in the other genome.
A group of alleles linked together on a chromosome from the same parent is called haplotype.
A paire alleles or a paire of haplotype is called genotype.
Alignment of WGS sample
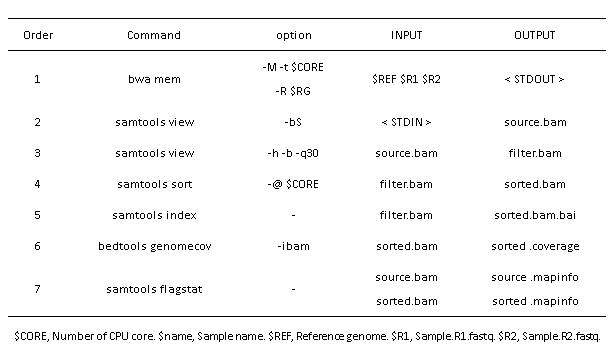
Each WGS sample clean reads were mapped to the reference genome using bwa-mem with the −M option.
Samtools(v1.8) was used to filter multiple mapping reads with -q30 option (mapping quality < 30) and to sort BAM files.
The Software and run order in the alignment script:
Call variation
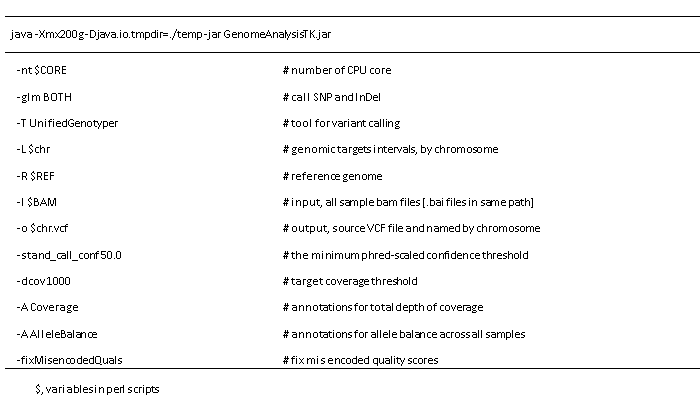
All sorted BAM files of one project were used for calling SNPs and indels by employed Genome Analysis Toolkit (GATK v3.8).
UnifiedGenotyper of GATK was used to generate VCF files, VariantFiltration of GATK was used to filter and annotate VCF files.
Options of UnifiedGenotyper:
Options of VariantFiltration:

Each project's VCF (filterd 'Low' annotation variation) files were loaded into MongoDB collections separately, genotype was generated with customized or default parameters by traverses all project collections.
Genotyping
Genotyping strategies based on multiple populations:
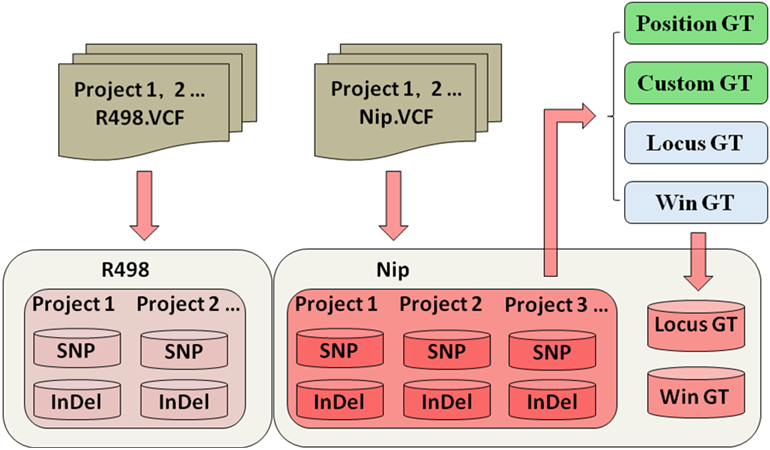
When query the Position GT and Custom GT, the system traverses all population project at backend, and generate the genotype in real-time.
After added a new population to the database, the Locus GT and Win GT needs to be regenerated at backend with offline, results of new genotype will be imported database for online query.